
Claudia Ctortecka was both sceptical and intrigued when her thesis adviser told her in 2018 about a new method that uses mass spectrometry to analyse the protein contents of individual cells. When he said he was looking for someone to pursue this single-cell proteomics strategy in his laboratory at the Vienna BioCenter research institute, she decided to take a chance.
“I was always very much interested in mass spectrometry,” she says. “And I thought, ‘why not go for the challenge?’ I wanted to look into that [strategy] a bit deeper and closer.”
With no backup plan, the project was a sink-or-swim proposition, Ctortecka says. “It was basically just: do single cell, make it work, or try harder.” Yet work it did. In April, she and her colleagues detailed a new sample-preparation device called proteoCHIP, which they used to map some 2,000 proteins across 158 single cells from 2 human cell types1.
That study is one of at least half a dozen over the past year that have described single-cell proteomics strategies, tools and preliminary findings. And more are coming. In 2018, Nikolai Slavov, a systems biologist at Northeastern University in Boston, Massachusetts, hosted his first annual conference on single-cell proteomics, which attracted about 50 attendees. This year’s (mostly virtual) conference had more than 1,300. “The growth has been exponential,” he says.
Most single-cell studies focus on nucleic acids, especially the transcriptome — which represents all the expressed genes in a cell. But proteins, says Neil Kelleher, a biochemist at Northwestern University in Evanston, Illinois, are “the worker bees” of the cell. “The amounts, the post-translational modifications, the proteoform dynamics — this is what is closer to the phenotype,” he explains. “And that means that disease diagnostics, response to drugs, all the human biology we want to engage with — to control, steer, detect — it needs proteomics.”

Single-cell analysis enters the multiomics age
Proteomics aims to catalogue and characterize the total complement of protein isoforms from a cell, tissue, organ or organism. (These ‘proteoforms’ are encoded by the same gene but have non-identical amino-acid sequences or post-translational modifications.) However, at the single-cell level, that’s easier said than done. Each type of nucleic acid behaves largely in a predictable way. But the proteome has a vast array of different chemistries, interactions, dynamics and abundances. And with no protein equivalent to PCR amplification of DNA, any technique to detect proteins must be sensitive enough to identify them, however little material a cell contains.
Using antibodies, that’s relatively straightforward. Flow cytometry and mass cytometry, for instance, can each quantify up to about 50 proteins per cell. And high-resolution microscopy, as used in the Human Protein Atlas project, intrinsically provides single-cell resolution.
But not all proteins have corresponding antibodies, and some antibodies bind to proteins only weakly or non-specifically. Furthermore, because antibody-based approaches target specific proteins, researchers can only see that portion of the proteome. Many in the single-cell proteomics community have instead turned to mass spectrometry, a non-targeted method that identifies and quantifies molecules on the basis of their mass and charge (see ‘Two paths to the proteome’).
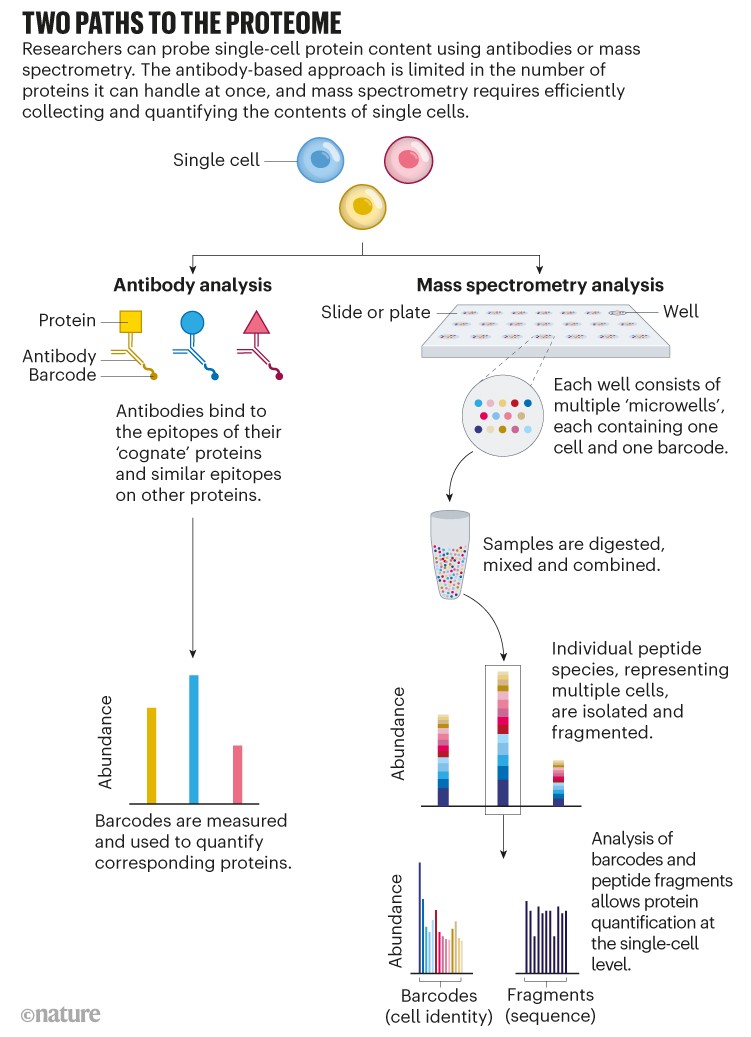
Source: Adapted from N. Slavov Science 367, 512–513 (2020).
The fact that mass spectrometry is sensitive enough to identify at least some proteins at the single-cell level was never in doubt: some instruments can detect attomolar (10–18 moles) quantities of material, the equivalent of several hundred thousand ions. According to one study2, the median mammalian protein is present at 18,000 copies per cell. But manipulating the contents of a single cell and faithfully transferring them into a mass spectrometer poses challenges.
As recently as five years ago, says Matthias Mann, director of the Max Planck Institute of Biochemistry in Munich, Germany, “the community was so far away from single-cell sensitivity, and also from handling single cells” that he used to think “it might happen some time, but not in my career”. Yet the field has accelerated faster than he expected.
Small samples
According to Mann, that acceleration stems not only from advances in instrumentation and analytical tools, but also, crucially, in sample preparation. “You want to have this whole reaction happen in a small volume so that you don’t lose the proteins and they don’t adsorb everywhere,” he explains.
Ctortecka’s proteoCHIP is one such design; another is nanoPOTS, developed by chemists Ryan Kelly and Ying Zhu at the Pacific Northwest National Laboratory in Richland, Washington.
NanoPOTS is like a nanolitre-scale microtiter plate fabricated onto the surface of a microscope slide3. Each ‘well’ is a hydrophobic circle about one millimetre in diameter, with a small hydrophilic ‘pedestal’ at the centre at which cells are deposited and prepared. “Think about the mesas in Arizona,” says Kelly, now at Brigham Young University in Provo, Utah, referring to the US state’s iconic, flat-topped hills: “All the stuff is taking place on the top of the mesa.”
The pedestal’s small area means there is a smaller surface for proteins to adhere to — about 99.5% less than a 0.5-millilitre centrifuge tube, as Kelly and Zhu note in their study. The correspondingly small reaction volumes (less than 200 nanolitres) increase enzyme concentration and thus efficiency. Add the fact that the reaction protocol limits liquid-handling steps, and the result is an increased yield of proteins per cell. Kelly’s team observed from 2- to 25-fold more peptides with nanoPOTS than when samples were prepared in 0.5-millilitre centrifuge tubes. Using nanoPOTS, Kelly’s team has detected an average of 1,085 and 1,012 proteins for each of two classes of primary human neuron4.

A dream of single-cell proteomics
How comprehensive that is depends on how you count — some genes encode multiple proteoforms, for instance, and not all proteins are expressed in all cells. However, that number is par for the course for single-cell proteomics: some researchers claim to have improved on it in unpublished work, but most studies identify about 1,000 proteins per cell (although the total number of identified proteins across all cells is higher). In a February preprint5, for instance, Mann’s team used a new instrument design from mass-spectrometry vendor Bruker in Billerica, Massachusetts, to detect proteome differences as cells progress through the cell cycle. The median number of proteins detected per cell-cycle stage ranged from 611 in the growth phase of cell division to 1,263 in the subsequent phase, when DNA is synthesized. Ongoing work has detected more (952 and 1,773, respectively). But that number was enough to tease apart biological differences. “Every single cell has quite a stable proteome,” Mann notes, meaning that researchers might be able to analyse fewer cells than other single-cell methods require. “Conceptually, that is the most exciting result of that paper,” Mann says.
It still takes a long time to acquire those data, however. Single-cell proteomics studies tend to use ‘bottom-up’ strategies to identify proteins from a smattering of peptide fragments rather than looking for intact proteins. But those peptides are identified one at a time, not in parallel. And the mass spectrometer needs time to accumulate each ion. For one study, Erwin Schoof, a biological mass spectrometrist at the Technical University of Denmark in Lyngby, allocated half a second per peptide in a 160-minute run. “On a good day we are measuring 4,500 peptides per cell,” Schoof says. As a result, his team could analyse just eight samples per day.
Sample preparation is also a bottleneck. With 27 wells, the original nanoPOTS could process 27 single cells at a time. Zhu’s second-generation ‘nested nanoPOTS’ (N2) design contains a 3 × 3 grid of pedestals in each well, supporting up to 243 cells (27 × 9) at once6. According to Zhu, N2 was designed to accommodate another crucial development in single-cell proteomics: multiplexing, which increases throughput.

Part of the mass spectrometer used by systems biologist Nikolai Slavov to study single-cell proteomics.Credit: Northeastern University/Ruby Wallau
In 2018, Slavov’s team described a method called SCoPE-MS (single-cell proteomics by mass spectrometry)7, which blends a mass-spectrometer-friendly cell-lysis protocol with a protein carrier that increases the amount of material available for sequencing. “This kind of approach immediately increased our ability to determine peptide sequences without doing anything difficult,” Slavov explains. “We were outsmarting the problem rather than brute-forcing it.”
Barcode breakthrough
Crucially, SCoPE-MS also features mass spectrometry’s version of barcoding: isobaric tags. These are molecules with identical masses that fragment into differently sized ions inside a mass spectrometer. By coupling different tags to different samples, researchers can work out how much of a given protein is present in each one. Using tandem mass tag (TMT) reagents, for instance, researchers can differentiate between up to 18 samples in a single mixture8. But to do so, the samples must be labelled individually and then pooled — a technically challenging step, given the small volumes involved. “The robot has to be very precise to withdraw this nanolitre volume and put them together for mass-spectrometer analysis,” Zhu explains. N2 allows researchers to process cells individually but then pool them in a single step by adding a large enough droplet of buffer to cover all the individual pedestals in one ‘well’, thus circumventing that issue.
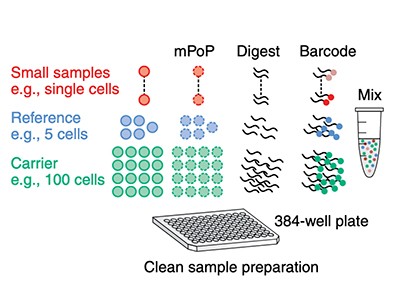
Towards resolving proteomes in single cells
At this year’s Single Cell Proteomics conference in Boston, Slavov’s graduate student Andrew Leduc presented an alternative approach. Leduc described how he and his colleagues used a piezo-acoustic dispensing device to array and prepare some 1,500 cells in 20-nanolitre droplets. These were clustered in groups of 12–14 on microscope slides to simplify pooling, and surrounded by a perimeter of water droplets to increase humidity and prevent evaporation9. The team has used that method to study macrophage stimulation and the cell division cycle.
Meanwhile, other members of Slavov’s team have revamped SCoPE-MS. SCoPE2 uses a simpler cell-lysis approach and improved analysis pipeline10, and is broadly accessible and scalable for production use, Slavov says.
Other researchers are trying to make the most of their instruments’ precious time by changing how they collect data. Most mass spectrometrists run their machines in a ‘data-dependent acquisition’ mode, in which the instrument identifies and sequences the most abundant ions. As a result, these analyses tend to overlook the most interesting, lower-abundance proteins.
Another option is a targeted approach, in which the instrument is told specifically which ions to look for. But some researchers are now exploring strategies that scan everything in the sample and work out the details later. These ‘data-independent analysis’ methods are not typically compatible with multiplexing, but in February, Ctortecka and her colleagues reported a strategy for combining the two11. “So you have a systematic way to look at your peptides in your sample, and this is performed in every single run exactly the same,” she says.
For his part, Schoof says he is working with vendors to accelerate chromatographic separations, and thus speed up experiments from 160 minutes to an hour. Using other optimizations, he has a roadmap to ramp up to 20 samples, or 360 multiplexed cells, per day. At that rate, he says, “a 10,000-cell experiment like you see in single-cell RNA-seq is, for lack of a better word, ‘only’ one month of runtime. In terms of doing single-cell proteomics, that’s already quite an achievement.”
Another dimension
Most single-cell methods remove cells from their tissue context. But where in the tissue a cell resides actually matters. By disaggregating cells, researchers lose what Mann calls their “sociology”. So he and other researchers are working to add a spatial dimension to single-cell proteomics, although none of the approaches is yet at the single-cell level.
Last year, Kelly and his colleagues published a strategy combining nanoPOTS, laser-capture microdissection (which uses a laser to excise cells from tissue) and mass spectrometry to detail some 2,000 proteins per 100-micrometre pixel12. In May, a team led by cancer researcher Thomas Cox of the Garvan Institute of Medical Research in Sydney, Australia, and vision scientist Gus Grey at the University of Auckland, New Zealand, combined ultra-high-resolution mass spectrometry and an R software package called HIT-MAP to sequence and identify proteins in intact samples of bovine lens tissue13.

NatureTech hub
And in January, Mann and his team reported a strategy called Deep Visual Proteomics14, which blends artificial intelligence, microscopy and laser-capture microdissection to automatically identify, isolate and characterize as few as 100 cells of a given type in tissue. His team used the approach to differentiate between cells at the centre and periphery of human melanoma samples. “I think this can be quite a game-changer,” he says.
Others, such as Kelleher, are pushing for single-molecule, single-cell proteomics — that is, the ability to sequence individual protein molecules in a cell. At the moment, he says, “we’re barely at proof-of-concept for some of these underlying technologies.” But their development is likely to get a boost. In July, the US National Institutes of Health announced some US$20 million in funding for technology development in single-molecule and single-cell proteomics. And Kelleher estimates that private investors have poured some $2 billion more into the subfield.
To make the most of those technologies, Kelleher and others advocate for a comprehensive atlas of all the human proteoforms that could be present in a sample. Just as the Human Genome Project provided a reference genome that made next-generation DNA sequencing technologies more powerful, Kelleher and his colleagues envision a Human Proteoform Project to create what they call “a definitive reference set of the proteoforms produced from the genome”15. Such a resource could enhance the power of both single-cell and single-molecule proteomics technologies by allowing researchers to concentrate more on ‘scoring’ proteins than discovering them, Kelleher says.
There’s no guarantee that such an atlas will come to pass. But when it comes to ’omics, one should never bet against the technology. When she started her doctoral work, Ctortecka doubted her project would succeed, but thought she would learn something interesting in any event. “I was very much convinced that this would never be possible,” she says. “Look where we are now.”
Nature 597, 580-582 (2021)
doi: https://doi.org/10.1038/d41586-021-02530-6
References
- Hartlmayr, D. et al. Preprint at bioRxiv https://doi.org/10.1101/2021.04.14.439828 (2021).
- Nagaraj, N. et al. Mol. Syst. Biol. 7, 548 (2011). PubMed Article Google Scholar
- Zhu, Y. et al. Nature Commun. 9, 882 (2018). PubMed Article Google Scholar
- Cong, Y. et al. Chem. Sci. 12, 1001–1006 (2021). Article Google Scholar
- Brunner, A.-D. et al. Preprint at bioRxiv https://doi.org/10.1101/2020.12.22.423933 (2021).
- Woo, J. et al. Preprint at bioRxiv https://doi.org/10.1101/2021.02.17.431689 (2021).
- Budnik, B., Levy, E., Harmange, G. & Slavov, N. Genome Biol. 19, 161 (2018). PubMed Article Google Scholar
- Li, J. et al. J. Proteome Res. 20, 2964–2972 (2021). PubMed Article Google Scholar
- Leduc, A., Huffman, R. G. & Slavov, N. Preprint at bioRxiv https://doi.org/10.1101/2021.04.24.441211 (2021).
- Specht, H. et al. Genome Biol. 22, 50 (2021). PubMed Article Google Scholar
- Ctortecka, C. et al. Preprint at bioRxiv https://doi.org/10.1101/2021.02.11.430601 (2021).
- Piehowski, P. D. et al. Nature Commun. 11, 8 (2020). PubMed Article Google Scholar
- Guo, G. et al. Nature Commun. 12, 3241 (2021). PubMed Article Google Scholar
- Mund, A. et al. Preprint at bioRxiv https://doi.org/10.1101/2021.01.25.427969 (2021).
- Smith, L. M. et al. Preprint at https://doi.org/10.20944/preprints202010.0368.v1 (2020).
